Rhabdoid Tumor: Causes, Symptoms, Diagnosis, Treatment & Prognosis
~Introduction
Rhabdoid tumor is a rare but highly aggressive form of cancer that primarily affects infants and very young children. First identified in the kidney, this tumor is now known to occur in multiple parts of the body, including the brain and soft tissues. Due to its rapid growth and early spread, rhabdoid tumor requires immediate medical attention and specialized treatment.
This detailed article explains everything you need to know about rhabdoid tumor — including its types, causes, symptoms, diagnosis, treatment options, survival rates, and ongoing research developments.
~What is a Rhabdoid Tumor?
A rhabdoid tumor is a fast-growing cancer made up of abnormal cells that appear “rhabdoid” (resembling muscle cells) under a microscope. Despite the name, these tumors do not arise from muscle tissue. Instead, they are linked to genetic mutations affecting tumor suppressor genes.
Rhabdoid tumors most commonly affect:
The kidneys
The brain
Soft tissues throughout the body
These cancers are most frequently diagnosed in children under the age of 3, although rare cases can occur in older children and adults.
~Types of Rhabdoid Tumors
1. Malignant Rhabdoid Tumor of the Kidney (MRTK)

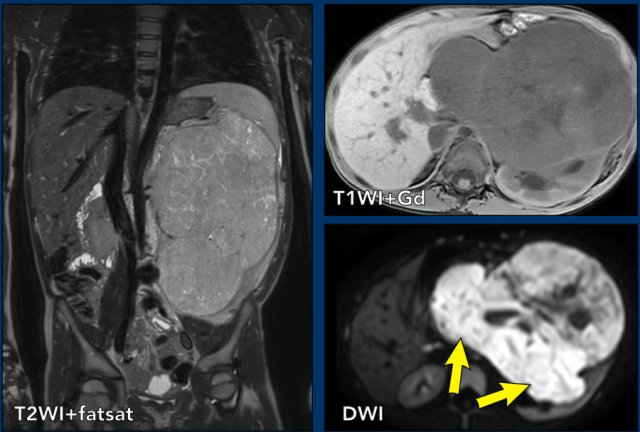


Malignant Rhabdoid Tumor of the Kidney (MRTK) was initially thought to be a variant of Wilms tumor but is now recognized as a distinct and more aggressive cancer. It primarily affects infants and toddlers.
It often presents as a large abdominal mass and may spread rapidly to the lungs or brain.
2. Atypical Teratoid/Rhabdoid Tumor (AT/RT)
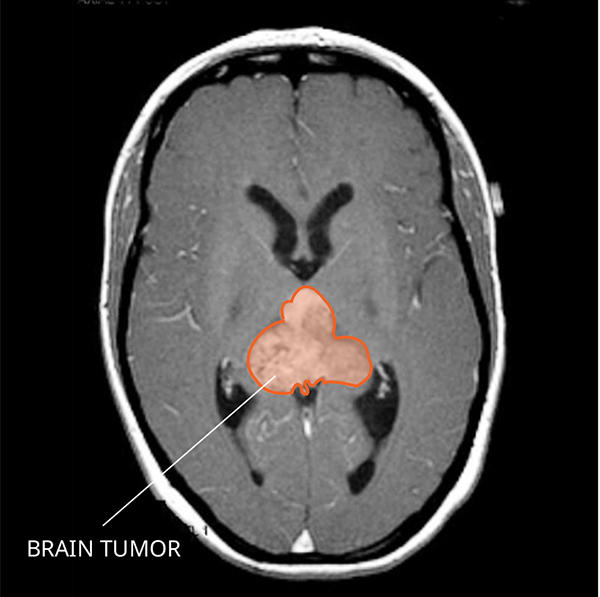


Atypical Teratoid/Rhabdoid Tumor (AT/RT) is a rhabdoid tumor that occurs in the central nervous system, most commonly in the cerebellum or brainstem. It is one of the most aggressive pediatric brain tumors.
Symptoms depend on tumor location and may include headaches, vomiting, or developmental delays.
3. Extrarenal Rhabdoid Tumor
These tumors develop outside the kidney and brain, often in soft tissues such as:
Neck
Liver
Chest
Pelvis
They are rare but equally aggressive.
~Causes and Genetic Factors
Rhabdoid tumors are strongly associated with mutations in the SMARCB1 gene, also known as the INI1 gene. This gene normally helps suppress tumor growth. When it is missing or mutated, uncontrolled cell division occurs.
Some children inherit this mutation, leading to a condition called Rhabdoid Tumor Predisposition Syndrome (RTPS). However, most cases occur sporadically without family history.
Key genetic aspects:
Loss of SMARCB1 gene function
Rarely, mutation in SMARCA4 gene
May be inherited or spontaneous
Genetic counseling is recommended for affected families.
~Risk Factors
Although rhabdoid tumors are rare, certain factors increase risk:
Age under 3 years
Genetic mutation in SMARCB1
Family history of rhabdoid tumor
Rhabdoid Tumor Predisposition Syndrome
There are no known environmental or lifestyle causes.
~Symptoms of Rhabdoid Tumor
Symptoms vary depending on tumor location.
Kidney Tumor Symptoms:
Abdominal swelling or mass
Blood in urine
Fever
Irritability
Weight loss
Brain Tumor (AT/RT) Symptoms:
Persistent headache
Vomiting
Balance problems
Seizures
Enlarged head size in infants
Soft Tissue Tumor Symptoms:
Swelling or lump
Pain
Breathing difficulty (if chest involved)
Because these tumors grow rapidly, symptoms often worsen quickly.
~Diagnosis of Rhabdoid Tumor
Early diagnosis is critical due to the tumor’s aggressive nature.
1. Physical Examination
Doctors check for lumps, neurological changes, or abdominal enlargement.
2. Imaging Tests
Ultrasound
CT scan
MRI
MRI is especially important for brain tumors.
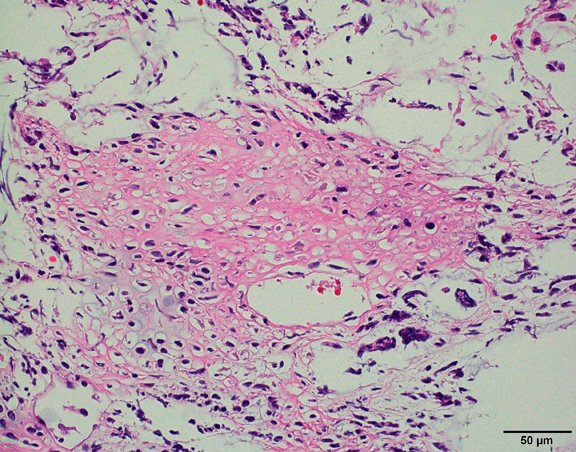
3. Biopsy
A tissue sample confirms the diagnosis. Pathologists look for:
Rhabdoid cell appearance
Loss of INI1 protein (key diagnostic marker)
4. Genetic Testing
Testing for SMARCB1 mutation helps confirm diagnosis and guide family counseling.
~Staging and Spread
Rhabdoid tumors are often advanced at diagnosis. They can spread (metastasize) to:
Lungs
Brain
Liver
Lymph nodes
Staging depends on:
Tumor size
Spread to distant organs
Lymph node involvement
Because of early metastasis, aggressive treatment is required.
~Treatment Options
Treatment is intensive and usually combines multiple therapies.
1. Surgery
Complete surgical removal of the tumor is the primary goal when possible.
Kidney removal (nephrectomy) for MRTK
Brain tumor resection for AT/RT
However, complete removal is not always achievable.
2. Chemotherapy
High-dose chemotherapy is standard treatment.
Common drugs include:
Vincristine
Cyclophosphamide
Doxorubicin
Cisplatin
Etoposide
Chemotherapy may be given before or after surgery.
3. Radiation Therapy
Used particularly in brain tumors, especially in older children. Radiation may be limited in infants due to long-term side effects.
4. Stem Cell Transplant
In some cases, high-dose chemotherapy followed by stem cell rescue is used to improve survival chances.
5. Targeted Therapy and Clinical Trials
Researchers are studying new treatments targeting genetic mutations. Immunotherapy and molecular-targeted drugs are under investigation.
Participation in clinical trials is often recommended.
~Prognosis and Survival Rate
Rhabdoid tumors are among the most aggressive childhood cancers.
Overall survival rates remain low, especially in:
Infants under 1 year
Metastatic disease
Incomplete tumor removal
However, survival has improved with aggressive multimodal therapy.
Early-stage diagnosis and complete surgical removal significantly improve outcomes.
~Complications
Possible complications include:
Tumor recurrence
Treatment-related side effects
Organ damage
Developmental delays (brain tumor cases)
Long-term follow-up is essential.
~Follow-Up and Survivorship
Children who survive require ongoing monitoring for:
Recurrence
Growth and development issues
Secondary cancers
Cognitive function (in brain tumor survivors)
Rehabilitation, educational support, and psychological counseling may be needed.
~Advances in Research
Recent research focuses on:
Targeting SMARCB1 mutation pathways
Epigenetic therapies
Precision medicine approaches
Improved chemotherapy protocols
Genomic studies are helping develop more personalized treatments.
~Emotional and Family Support
A diagnosis of rhabdoid tumor is devastating for families. Support resources include:
Pediatric oncology teams
Genetic counselors
Support groups
Palliative care specialists
Emotional and psychological care is crucial alongside medical treatment.
~Prevention and Screening
There is no known way to prevent rhabdoid tumors.
However, children with known SMARCB1 mutations may undergo:
Regular MRI scans
Routine physical exams
Early detection may improve survival.
~Frequently Asked Questions (FAQs)
1. Is rhabdoid tumor hereditary?
Most cases are not inherited, but some are linked to genetic mutations passed down in families.
2. What age is most affected?
Children under 3 years are most commonly diagnosed.
3. Is rhabdoid tumor curable?
It can be treated, but it is aggressive and survival depends on early diagnosis and response to therapy.
4. How rare is rhabdoid tumor?
It is extremely rare, accounting for a small percentage of pediatric cancers.
5. Can adults get rhabdoid tumors?
Rarely, but most cases occur in infants and toddlers.
~Conclusion
Rhabdoid tumor is a rare and highly aggressive childhood cancer that requires urgent, specialized treatment. Whether occurring in the kidney, brain, or soft tissues, it is linked to genetic mutations and tends to spread quickly.
Although prognosis remains challenging, advances in surgery, chemotherapy, stem cell therapy, and genetic research are improving outcomes. Early detection, comprehensive treatment, and strong family support play vital roles in managing this condition.




No comments:
Post a Comment